Sunday, September 25, 2016
Thursday, September 22, 2016
Treatment of Iron Deficiency - Risks
Iron overdose as a cause of a high anion gap metabolic acidosis
Now, iron is usually mentioned as an important cause of metabolic acidosis, and there is a warm spot reserved for it in the “MUDPILES” mnemonic. An impressionable person might be inclined to believe that iron contributes to the high anion gap metabolic acidosis by dissociating into unmeasured anions, much like the toxic alcohols. However, that would be wildly inaccurate, because iron is a cation.
The acidosis here is multifactorial. Some textbooks (Fowler’s Handbook on the Toxicology of Metals) suggest that the acidosis is mainly due to the physicochemical effects of the iron ion itself. Other sources (Goldfranks Manual of Toxicologic Emergencies) attribute the acidosis to a raised lactate, of which not all is generated by direct effects of the iron, but rather due to the fluid loss (from an ulcerated gut), cardiogenic shock (due to the myocardial mitochondrial toxicity) and fulminant hepatic failure. On top of that, a fair portion of the lactic acidosis is due to the direct mitochondrial toxicity of iron in all tissues.

Monday, September 19, 2016
Sensitivity, Specificity, Positive Predictive Value, and Negative Predictive Value
Sensitivity, Specificity, Positive Predictive Value, and Negative Predictive Value
 In this example, two columns indicate the actual condition of the subjects, diseased or non-diseased. The rows indicate the results of the test, positive or negative.
In this example, two columns indicate the actual condition of the subjects, diseased or non-diseased. The rows indicate the results of the test, positive or negative.
Cell A contains true positives, subjects with the disease and positive test results. Cell D subjects do not have the disease and the test agrees.
A good test will have minimal numbers in cells B and C. Cell B identifies individuals without disease but for whom the test indicates 'disease'. These are false positives. Cell C has the false negatives.
If these results are from a population-based study, prevalence can be calculated as follows:
- Prevalence of Disease= Tdisease/ Total × 100
The population used for the study influences the prevalence calculation.
Sensitivity is the probability that a test will indicate 'disease' among those with the disease:
- Sensitivity: A/(A+C) × 100
Specificity is the fraction of those without disease who will have a negative test result:
- Specificity: D/(D+B) × 100
Sensitivity and specificity are characteristics of the test. The population does not affect the results.
A clinician and a patient have a different question: what is the chance that a person with a positive test truly has the disease? If the subject is in the first row in the table above, what is the probability of being in cell A as compared to cell B? A clinician calculates across the row as follows:
- Positive Predictive Value: A/(A+B) × 100
- Negative Predictive Value: D/(D+C) × 100
Positive and negative predictive values are influenced by the prevalence of disease in the population that is being tested. If we test in a high prevalence setting, it is more likely that persons who test positive truly have disease than if the test is performed in a population with low prevalence..
Let's see how this works out with some numbers...
Hypothetical Example 1 - Screening Test A
 100 people are tested for disease. 15 people have the disease; 85 people are not diseased. So, prevalence is 15%:
100 people are tested for disease. 15 people have the disease; 85 people are not diseased. So, prevalence is 15%:- Prevalence of Disease:
Tdisease/ Total × 100,
15/100 × 100 = 15%
Sensitivity is two-thirds, so the test is able to detect two-thirds of the people with disease. The test misses one-third of the people who have disease.
- Sensitivity:
A/(A + C) × 100
10/15 × 100 = 67%
The test has 53% specificity. In other words, 45 persons out of 85 persons with negative results are truly negative and 40 individuals test positive for a disease which they do not have.
- Specificity:
D/(D + B) × 100
45/85 × 100 = 53%
The sensivity and specificity are characteristics of this test. For a clinician, however, the important fact is among the people who test positive, only 20% actually have the disease.
- Positive Predictive Value:
A/(A + B) × 100
10/50 × 100 = 20%
For those that test negative, 90% do not have the disease.
- Negative Predictive Value:
D/(D + C) × 100
45/50 × 100 = 90%
Now, let's change the prevalence..
Hypothetical Example 2 - Increased Prevalence, Same Test
This time we use the same test, but in a different population, a disease prevalence of 30%.
 Prevalence of Disease:
Prevalence of Disease:
Tdisease/ Total × 10
30/100 × 100 = 30%
We maintain the same sensitivity and specificity because these are characteristic of this test.
- Sensitivity:
A/(A + C) × 100
20/30 × 100 = 67%
- Specificity:
D/(D + B) × 100
37/70 × 100 = 53%
Now let's calculate the predictive values:
- Positive Predictive Value:
A/(A + B) × 100
20/53 × 100 = 38%
- Negative Predictive Value:
D/(D + C) × 100
37/47 × 100 = 79%
Using the same test in a population with higher prevalence increases positive predictive value. Conversely, increased prevalence results in decreased negative predictive value. When considering predictive values of diagnostic or screening tests, recognize the influence of the prevalence of disease. The figure below depicts the relationship between disease prevalence and predictive value in a test with 95% sensitivity and 95% specificity:

Relationship between disease prevalence and predictive value in a test with 95% sensitivity and 85% specificity.
(From Mausner JS, Kramer S: Mausner and Bahn Epidemiology: An Introductory Text. Philadelphia, WB Saunders, 1985, p. 221.)
(From Mausner JS, Kramer S: Mausner and Bahn Epidemiology: An Introductory Text. Philadelphia, WB Saunders, 1985, p. 221.)
 Think About It!
Think About It!
Come up with an answer to this question and then click on the icon to the left to reveal the answer.
Under what circumstance would you really want to minimize the false positives?
answer: Minimizing false positives is important when the costs or risks of followup therapy are high and the disease itself is not life-threatening...prostate cancer in elderly men is one example; as another, obstetricians must consider the potential harm from a false positive maternal serum AFP test (which may be followed up with amniocentesis, ultrasonography and increased fetal surveillance as well as producing anxiety for the parents and labeling of the unborn child), against potential benefit.
Think About It!
Come up with an answer to this question and then click on the icon to the left to reveal the answer.
When would you want to minimize the false negatives?
answer: We don’t want many false negative if the disease is often asymptomatic and
- is serious, progresses quickly and can be treated more effectively at early stages OR
- easily spreads from one person to another
What is a good test in a population? Actually, all tests have advantages and disadvantages, such that no test is perfect. There is no free lunch in disease screening and early detection.
Thursday, September 15, 2016
Cramps
Cramps
Causes
Overuse of a muscle, dehydration, muscle strain or simply holding a position for a prolonged period can cause a muscle cramp. In many cases, however, the cause isn't known - idiopathic.
Although most muscle cramps are harmless, some may be related to an underlying medical condition, such as:
- Inadequate blood supply. Narrowing of the arteries that deliver blood to your legs (arteriosclerosis of the extremities) can produce cramp-like pain in your legs and feet while you're exercising. These cramps usually go away soon after you stop exercising.
- Nerve compression. Compression of nerves in your spine (lumbar stenosis) also can produce cramp-like pain in your legs. The pain usually worsens the longer you walk. Walking in a slightly flexed position — such as you would use when pushing a shopping cart ahead of you — may improve or delay the onset of your symptoms.
- Mineral depletion. Too little potassium, calcium or magnesium in your diet can contribute to leg cramps. Diuretics — medications often prescribed for high blood pressure — also can deplete these minerals.
Neuromuscular Cramps

Possible roles of calcium and ATP in muscle cramps. Too little ATP means calcium stays in the sarcoplasma and muscles stay contracted. ischemia = low oxygen = low ATP

Friday, September 9, 2016
Practice Questions - Cell Electrophysiology
the link below is to a set of practice questions on
cell electrophysiology - this is updated with more questions and scoring (9/25/16)
Cell Physiology Practice Questions
after you do the practice questions you submit your answers and you will see your results as well as a summary of the responses of students who have taken the quiz.
There is also a link to an explanation of the answers
cell electrophysiology - this is updated with more questions and scoring (9/25/16)
Cell Physiology Practice Questions
after you do the practice questions you submit your answers and you will see your results as well as a summary of the responses of students who have taken the quiz.
There is also a link to an explanation of the answers
Case study; 17 y/o male with muscle weakness - Case and answers
17 y/o male with muscle weakness
Case Authors: Steve Wood, PhD
Tracey Milligan, MD
Case Based Learning
You will work on this case in 8 groups of 7 students per group. The group should discuss all the questions (10 min) and then each member of the group should pick one of the 7 questions to research during the during the next 30 minutes of the first session and submit their answer using this form. During the last 10 minutes of the first session, the 7 students who researched each question will form a new group (e.g., 7 students who worked on question 1) and take 10 minutes to discuss their individual answers and make plans for coming up with a group consensus answer to be presented during the second session for the case. During the second session, one or more students from each group will present the answer to their group’s question for 5 minutes followed by 2 minutes for questions from the class.
Learning Objectives
- Draw a concept map and explain the mechanisms (hormonal, renal, cellular) involved in potassium homeostasis.
- Draw and label the phases of an action potential for a skeletal muscle cell, cardiac muscle cell, and sinoatrial node.
- Describe the effects of hypokalemia and hyperkalemia on the resting membrane potential and potassium conductance of cells including nerve and muscle.
- Describe how surreptitious use of diuretics can cause hypokalemia and explain how one diuretic (acetazolamine) can be used to treat hypokalemia.
- Explain the mechanisms of muscle weakness in hypokalemia.
- Describe the genetics and molecular mechanisms of hypokalemic periodic paralysis.
- Explain the mechanisms that lead to transcellular shifts of potassium. Describe how marijuana intoxication may lead to hypokalemia.
- Bonus question (anybody can do this one): Explain the abnormal findings on the EKG.
Pre-study
Strong Medicine - video
Gordon Hu is a 17-year-old member of his high school wrestling team. He is 5’ 10” tall and competes in the 145 - 150 lb. division. He struggles to make this weight for matches as his weight is normally 160 lbs. He is extremely muscular and exercises frequently outside of practice including cardio and strength training. He doesn’t drink or smoke cigarettes but does smoke marijuana with his friends.
He knew it was normal to be tired and feel weak after a hard practice or match, but lately he had noticed extreme weakness and his legs felt “like rubber”. He thought that maybe he had low blood sugar so he made it a point to do some “carb loading” before a match. Instead of improving his symptoms, this seemed to make them worse. After his most recent match, he had to carried off the mat after he was pinned, suffering his 3rd straight loss. After he collapsed, Gordon was terrified when he discovered that although he was conscious, he couldn’t open his eyes or speak for about 30 seconds; then he was o.k. but still had to be helped off the mat.
Gordon’s parents were at the wrestling match and were very alarmed at what had happened. His father remembered having similar problems when he was a high school athlete. They told Gordon they were going to make an appointment with the family doctor. The next morning, they met with Dr. Rhodes, their family physician. Dr. Rhodes talked to Gordon about his problem and made the following notes on Gordon’s chart:
Gordon’s symptoms were recent onset. He did not have any problems like this when he was a child. He started noticing some weakness after he went out for wrestling in 10th grade (2 years ago). The problem only occurred once in awhile. Sometimes he would have no problems for 6 months, and then experience weakness several times a month. Gordon has no siblings. He reports having several girlfriends and is sexually active. No recent travel. Childhood illnesses were chickenpox, mumps, and measles. Physical exams have been normal. Denies cigarette and alcohol use. He smokes marijuana “occasionally”. Due to physique, inquiry was made re: steroid use. Pt. denies using any steroids. Gordon’s father reported similar problems when he was involved in high school athletics. He remembered that some “supplement” prescribed by the family doctor made it better.
Dr. Rhodes took a blood sample and sent it to the lab next door for a CBC and electrolyte panel. He also took a muscle biopsy for testing. While waiting for the results he did a physical exam:
Physical Exam
General - healthy young man, alert, oriented.
Vital signs
Oxygen saturation 98%
Blood pressure 120/80 mm Hg
Pulse 66 beats per minute
Respiration 12 bpm
Weight 160 lbs
Height 5 ft. 10 in
Temperature 98.6 F
Laboratory Tests/Investigations
CBC
Hb 15 g/dL (normal 14-18 g/dL)
Hct 45 % (normal 42-52%)
White blood count (WBC) 12 x 103 (normal 5-10 x 103/mm3)
Neutrophils – elevated
Lymphocytes – nl
Monocytes - nl
Eosinophils – nl
Basophils - nl
Platelets 450,000 (normal 150,000 – 400,000/mm3)
Electrolytes
Na+ - 135 (normal 135 – 145 mEq/L)
K+ - 4.4 (normal 3.5 – 5 mEq/L)
Cl- - 110 (normal 100 – 110 mEq/L)
Because the test results were mostly normal, Dr. Rhodes decided to repeat the blood test after Gordon had done 40 pushups in the office. The electrolyte results were:
Electrolytes
Na+ 135 (normal 135 – 145 mEq/L)
K+ 2.2 (normal 3.5 – 5 mEq/L)
Cl- 110 (normal 100 – 110 mEq/L)
A 12 lead EKG was obtained because of the low K+


Normal EKG
Dr. Rhodes told Gordon that he has a condition called hypokalemic periodic paralysis. He explained that there is no cure but it can be treated. He explained that it is an inherited trait due to a gene mutation.
Discuss the following questions/learning issues and each member pick 1 of them for presentation during the second session. Submit your answers using this form.
- Draw a concept map and explain the mechanisms (hormonal, renal, cellular) involved in potassium homeostasis.
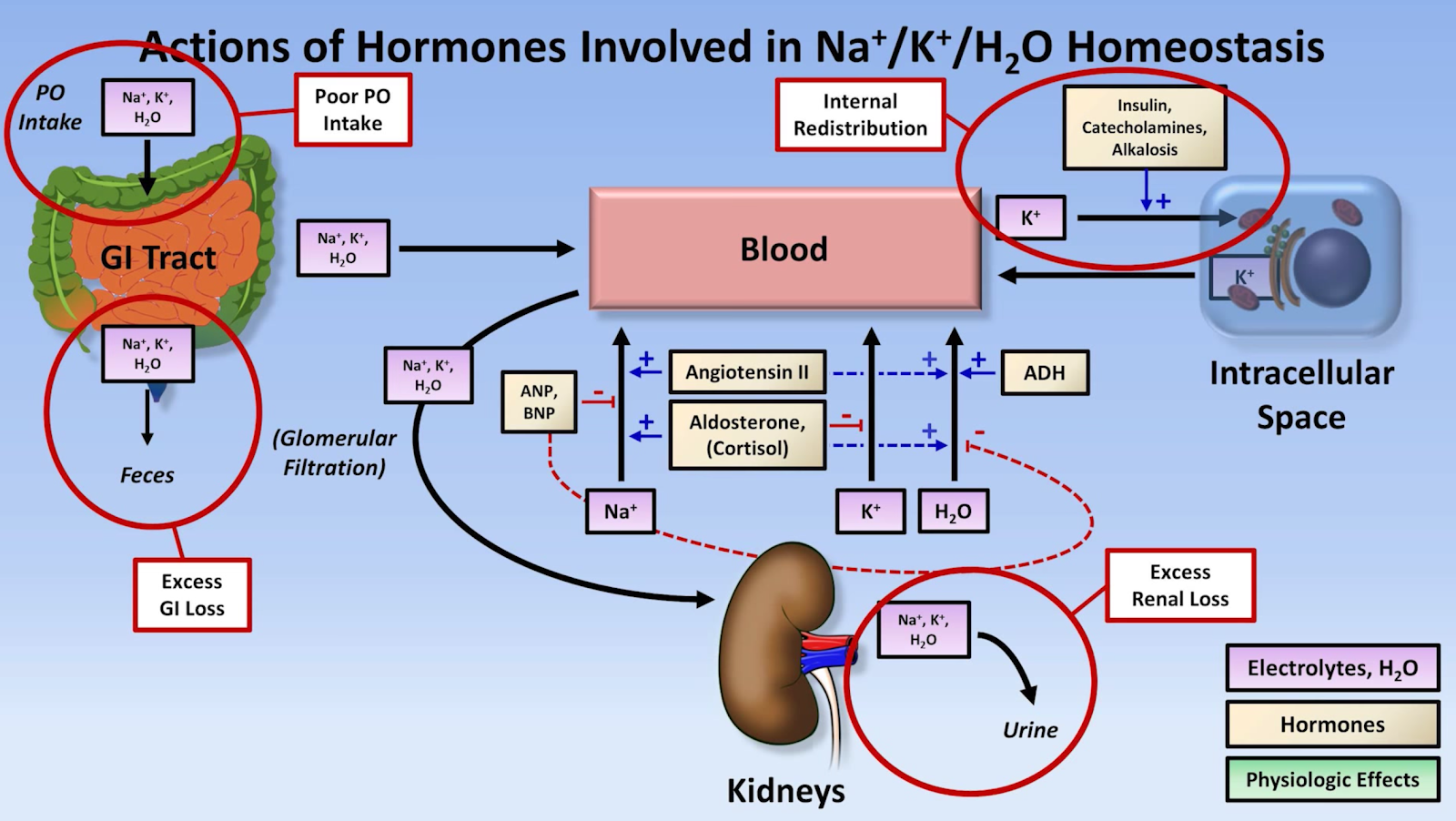
Also, to amplify the renin-angiotensin-aldosterone system:

High potassium is one of the signals that stimulates production of renin which catalyzes the conversion of angiotensinogen to angiotensin I to start the signalling pathway.
2. Draw and label the phases of an action potential for a neuron, skeletal muscle cell, cardiac muscle cell, and sinoatrial node. 





3. Describe the effects of hypokalemia and hyperkalemia on the resting membrane potential and potassium conductance of cells including nerve and muscle.
Hyperkalemia effects on cardiac muscle action potentials are summarized in this figure: During phase 4, the resting membrane potential is depolarized (less negative) according to the Nernst equation

Phase 0 of the action potential occurs when voltage gated sodium channels open and sodium enters the myocyte down its electrochemical gradient. The rate of rise of phase 0 of the action potential (Vmax) is directly proportional to the value of the resting membrane potential at the onset of phase 0. This is because the membrane potential at the onset of depolarization determines the number of sodium channels activated during depolarization, which in turn determines the magnitude of the inward sodium current and the Vmax of the action potential.
Hyperkalemia also has profound effects upon phase 2 and phase 3 of the action potential. After the rapid influx of sodium across the cell membrane in phase 0, potassium ions leave the cell along its electrochemical gradient, which is reflected in phase 1 of the action potential. As the membrane potential reaches –40 to –45 mV during phase 0, calcium channels are stimulated, allowing calcium to enter the myocyte. The maximum conductance of these channels occurs approximately 50 msec after the initiation of phase 0 and is reflected in phase 2 of the action potential.
During phase 2, potassium efflux and calcium influx offset one another so that the electrical charge across the cell membrane remains the same, and the so-called plateau phase of the action potential is created (Fig. 3). During phase 3, the calcium channels close, while the potassium channels continue to conduct potassium out of the cell; in this way, the electronegative membrane potential is restored.7 One of the potassium currents (Ikr), located on the myocyte cell membrane, is mostly responsible for the potassium efflux seen during phases 2 and 3 of the cardiac action potential.10 For reasons that are not well understood, these Ikr currents are sensitive to extracellular potassium levels, and as the potassium levels increase in the extracellular space, potassium conductance through these currents is increased so that more potassium leaves the myocyte in any given time period.10 This leads to an increase in the slope of phases 2 and 3 of the action potential in patients with hyperkalemia and therefore, to a shortening of the repolarization time. This is thought to be the mechanism responsible for some of the early electrocardiographic manifestations of hyperkalemia, such as ST-T segment depression, peaked T waves, and Q-T interval shortening.

As illustrated in Figure 4, Vmax is greatest when the resting membrane potential at the onset of the action potential is approximately –75 mV, and does not increase as the membrane potential becomes more negative. Conversely, as the resting membrane potential becomes less negative (that is, –70 mV), as in the setting of hyperkalemia (Fig. 3), the percentage of available sodium channels decreases. This decrease leads to a decrement in the inward sodium current and a concurrent decrease in the Vmax; therefore, as the resting membrane potential becomes less negative in hyperkalemia, Vmax decreases. This decrease in Vmax causes a slowing of impulse conduction through the myocardium and a prolongation of membrane depolarization; as a result, the QRS duration is prolonged.
Hypokalemia would hyperpolarize the resting membrane potential but this does not change the slope of phase 0 as the Vmax does not change (Fig. 4).
Causes of Hyperkalemia
Numerous causes of hyperkalemia are seen in clinical practice. The most common are renal disease and the ingestion of medications that predispose the patient to hyperkalemia.2 Medications known to cause hyperkalemia include angiotensin-converting enzyme inhibitors, angiotensin-receptor blockers, penicillin G, trimethoprim, spironolactone, succinylcholine, alternative medicines, and heparin.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1413606/pdf/20060300s00010p40.pdf
4. Describe how surreptitious use of diuretics can cause hypokalemia and explain how one diuretic (acetazolamine) can be used to treat hypokalemia.
CA inhibitors, like acetylzolamide, are effective in many cases of Hypokalemic periodic paralysis. This is at first a paradox because CA inhibitors cause potassium loss and hypokalemia. However, CA inhibitors also impair bicarbonate reabsorption in the renal tubules. The resulting urinary loss of bicarb causes metabolic acidosis. The increased hydrogen ion causes a shift of potassium out of cells, alleviating the hypokalemia. Also results in less Ca++ binding to Protein-, increasing contractability.
5. Explain the mechanisms of muscle weakness in hypokalemia.
- Low potassium causes vasoconstriction and reduced muscle blood flow, leading to lactic acid accumulation in muscles. This causes osmotic entry of water which impairs calcium channels. Reduced calcium movement impairs contraction.
- Hypokalemia will decrease potassium channel conductance, which will lengthen repolarization time of a nerve cell. If this gets to be severe enough, transmission of action potentials will be disrupted, and the result can be generalized weakness or paralysis because signaling to the muscles are disrupted.
Hyperkalemia also causes muscle weakness:
It might be expected that this depolarization would make it easier to generate action potentials in the muscle because the resting membrane potential would be closer to threshold. A more important effect of depolarization, however, is that it closes the inactivation gates on Na + channels. When these inactivation gates are closed, no action potentials can be generated, even if the activation gates are open. Without action potentials in the muscle, there can be no contraction.
6. Describe the genetics and molecular mechanisms of hypokalemic periodic paralysis.
The physiologic basis of flaccid weakness is inexcitability of the muscle membrane (ie, sarcolemma). Alteration of serum potassium level is not the principal defect in primary PP; the altered potassium metabolism is a result of the PP. In primary and thyrotoxic PP, flaccid paralysis occurs with relatively small changes in the serum potassium level, whereas in secondary PP, serum potassium levels are markedly abnormal.
The sodium channel has 2 gates (activation and inactivation) and can exist in 3 states. At rest with the membrane polarized, the activation gate is closed and the inactivation gate is opened. With depolarization, the activation gate opens, allowing sodium ions to pass through the ion channel and also exposing a docking site for the inactivation gate. With continued depolarization, the inactivation gate closes, blocking the entry of sodium into the cell and causing the channel to enter the fast-inactivation state. This inactivation of the channel allows the membrane to become repolarized, resulting in a return to the resting state with the activation gate closed and the inactivation gate opened. Two inactivation processes occur in mammalian skeletal muscle: Fast inactivation involves terminating the action potential and acts on a millisecond time scale. Slow inactivation takes seconds to minutes and can regulate the population of excitable sodium channels.
Sodium channel mutations that disrupt fast and slow inactivation are usually associated with a phenotype of HyperPP and myotonia, where as mutations that enhance slow or fast inactivation producing loss of sodium channel function cause HypoPP.
some forms of HypoPP involve mutations of calcium and/or potassium channels.

7. Explain the mechanisms that lead to transcellular shifts of potassium. Describe how marijuana intoxication may lead to hypokalemia.
Electrolyte abnormalities reported in marijuana users contribute to pathology. Chronic marijuana users have lower serum sodium and potassium than non-users.[17] The heavy consumption of carbohydrates while intoxicated leads to an increase in serum insulin levels, driving potassium into cells and causing serum hypokalemia.[18]
The heavy consumption of carbohydrates while intoxicated leads to an increase in serum insulin levels, driving potassium into cells and causing serum hypokalemia.[18] This hypokalemia can produce reentrant arrhythmias by decreasing conductivity and increasing the resting membrane potential, duration of the action potential, and duration of the refractory period.[19] EKG changes include the decrease in T-wave amplitude, presence of U waves and a prolonged QTc.
Memory tool: Al Klow sis

8. Bonus question (anybody can do this one): Explain the abnormal findings on the EKG.
This hypokalemia can produce reentrant arrhythmias by decreasing conductivity and increasing the resting membrane potential, duration of the action potential, and duration of the refractory period.[19] EKG changes include the decrease in T-wave amplitude, presence of U waves and a prolonged QTc.

The decrease in Vmax in hyperkalemia causes a slowing of myocardial conduction, manifested by progressive prolongation of the P wave, PR interval, and QRS complex.
The maximum conductance of these channels occurs approximately 50 msec after the initiation of phase 0 and is reflected in phase 2 of the action potential. During phase 2, potassium efflux and calcium influx offset one another so that the electrical charge across the cell membrane remains the same, and the so-called plateau phase of the action potential is created (Fig. 3). During phase 3, the calcium channels close, while the potassium channels continue to conduct potassium out of the cell; in this way, the electronegative membrane potential is restored.7 One of the potassium currents (Ikr), located on the myocyte cell membrane, is mostly responsible for the potassium efflux seen during phases 2 and 3 of the cardiac action potential.10 For reasons that are not well understood, these Ikr currents are sensitive to extracellular potassium levels, and as the potassium levels increase in the extracellular space, potassium conductance through these currents is increased so that more potassium leaves the myocyte in any given time period.10 This leads to an increase in the slope of phases 2 and 3 of the action potential in patients with hyperkalemia and therefore, to a shortening of the repolarization time. This is thought to be the mechanism responsible for some of the early electrocardiographic manifestations of hyperkalemia, such as ST-T segment depression, peaked T waves, and Q-T interval shortening.


Subscribe to:
Posts (Atom)