This post contains email questions and my answers
A Malpighian body has two components- the glomerulus (a tuft of capillaries formed by the afferent arteriole) and the Bowman's capsule (a double-walled epithelial cup that encloses the glomerulus. Filtration occurs if the outward driving pressures exceed the inward reabsorption pressures. This balance is quantified by the Starling equation.
- Overview
- As mentioned, the rate of glomerular filtration is essentially governed by Starling Forces. However, because the glomerular capillaries are surrounded by the fluid in the Bowman's Capsule, the hydrostatic and oncotic pressure of Bowman's Space is used instead of those of the 'Interstitial Fluid'. Importantly, because no plasma proteins can cross the glomerular barrier during glomerular filtration, the oncotic pressure of fluid in Bowman's Space is essentially zero and is thus removed from the starling equation.
- Formally:
- GFR = Kf [(Pc-Pb)-(Πc)]
- GFR = Glomerular Filtration Rate (ml/min). Equivalent of Jv in Starling Forces
- Kf = Permeability Constant of glomerular capillaries
- Pc = Glomerular Capillary Hydrostatic Pressure
- Pb = Bowman's Space Hydrostatic Pressure
- Πc = Glomerular Capillary Oncotic Pressure
Physical determinants of the glomerular filtration rate
The Glomerular Filtration Rate (GFR) is a quantitation of the volume of fluid filtered through the glomerular barrier per unit time. The GFR is essentially determined by a special case of the starling equation and is given above. The primary physiological modulators of GFR are the afferent and efferent arteriolar resistances (RA and RE). As expected, increasing the afferent arteriolar resistance (RA) drops the pressure within the glomerulus and thus reduces GFR. The relationship between the efferent arteriolar resistance (RE) and GFR is more complicated. Initially, increasing RE boosts the pressure within the glomerulus and thus increases GFR. However, at higher values of RE the total blood flow through the glomerulus decreases and thus GFR drops.
I think you meant homeostasis instead of hemostasis. Homeostasis is the concept of animals maintaining a constant internal environment even when the external environment and/or stress factors or injury are present. Adaptation refers to how homeostasis is accomplished. Two examples:
1 Blood pressure is maintained within a narrow range of values even when there is a sudden change in gravity; e.g., when you rapidly stand up. The mechanisms that allow this adaptation are a baroreceptor reflex (rapid) and hormonal changes driven by renin produced in the kidneys (slow).
2 Arterial oxygen content is maintained at a normal value (20 mlO2/dL blood) even after you move from sea level to high altitude. The mechanisms that allow this adaptation are increased lung ventilation (rapid) and increased red blood cells driven by erythropoietin produced by the kidneys (slow).
Blood vessels

Hi Dr. Wood,
Could you please explain why there is oliguria in Nephritic syndrome?

Mechanisms of oliguria and acute kidney injury. Multiple mechanisms can potentially cause oliguria in the acutely injured kidney. Regional intrarenal differences in blood flow and redistribution; glomerular injury or altered intraglomerular hemodynamics; impaired tissue oxygenation causing preferential ischemia to the S 3 segment of the proximal convoluted tubule and the oxygen-avid thick ascending loop of Henle; loss of osmolar gradient, interstitial edema, or inflammation; and tubular or lower urinary tract obstruction can precipitate oliguria. CCD and MCD, cortical and medullary collecting ducts; Osm, osmolality; p t O 2 , tissue partial pressure of oxygen; Q B , renal blood flow; S 1 – S 3 , segments of the proximal convoluted tubule; TALH, thick ascending loop of Henle.
We are in MSK but I’ve come across a topic I’m lost in. I’m going over NSAID pharmacology and am struggling to understand the mechanism behind hyperkalemia associated with their use. From my understanding, the use of NSAIDs will block dilation of the renal afferent arteriole which would lead to a drop in GFR. I was under the impression that the kidney would sense this and would subsequently hype up the RAAS which would increase sodium and excrete potassium and H+ ions. However, this is clearly not the case as you see a Hyper- rather than Hypo- kalemia with certain NSAID use.
Do you happen to know the mechanism behind this?
- Author:
- Richard H Sterns, MD
- Section Editor:
- Michael Emmett, MD
- Deputy Editor:
- John P Forman, MD, MSc
In pulmonary arteries, hypoxia causes a triphasic response ending with a sustained phase of contraction52 and is termed “hypoxic pulmonary vasoconstriction”. This phenomenon is observed in isolated systemic or pulmonary arteries across many species including reptiles, birds and mammals53,54. Its physiological role is to adjust the ventilation/perfusion ratio by diverting blood to regions of the lung with adequate oxygen supply55. However sustained hypoxic constrictions may lead to pulmonary hypertension; the latter is a dramatic pathological situation still in search of an appropriate cure55,56. The acute phase of hypoxic constriction of pulmonary blood vessels is endothelium-dependent57,58, and ultimately results from the activation of Rho kinase leading to calcium sensitization in the vascular smooth muscle59. It is enhanced by inhibitors of NOS and phosphodiesterase V60. Metallothionein, a protein responsible for zinc homeostasis, may also be involved, since zinc chelation inhibits the contractile response in pulmonary arteries61. As regards the sustained phase of hypoxic constriction in the pulmonary circulation, cyclic adenosine diphosphateribose (cADPR), a molecule that modulates the activity of ryanodine receptors, appears to be responsible62. Indeed, hypoxia increases the production of cADPR, which facilitates calcium release from the sarcoplasmic reticulum63. Furthermore, the cADPR antagonist, 8-bromo-cADPR, abolishes the sustained phase of the hypoxic response64. These changes work in conjunction with calcium sensitization (also induced by hypoxia51) to produce hypoxic constriction.
1) If you’re sleeping and your heart rate drops to say, the 40’s, is the SA node still the starting point for depolarization or does the AV start to play a role? I would assume the drop in HR is due to a decrease in conduction velocity and that the SA node is still the primary depolarizer.
2) An athletic heart beats slower due to the increased stroke volume, which is due to strengthening of the left ventricle. At what point does LVH become pathologic? Are extreme athletes more prone to left heart problems?
Is the sum of partial pressures of gases in blood equal to total atmospheric pressure?
Not necessarily. This is true for alveolar gas pressures but not for blood. For practical purposes, liquids are incompressible and do not respond to changes in ambient pressure. Because of this, the sum of gas partial pressures in a liquid can be less than ambient pressure. Liquids such as blood and other body tissues will equilibrate only with the gas partial pressures to which they are exposed.
On the alveolar side of the alveolar membrane, the total partial pressures must equal ambient pressure. However, on the liquid side of the membrane, the total partial pressures can be less, and in some areas may be quite a bit less than ambient pressure. The partial pressure that a gas exerts in a liquid depends on the temperature, the solubility of the gas in the liquid and the amount of gas present. Thus, if the amount of gas present and the temperature remain constant, the partial pressure of the gas in a tissue is fixed. If one gas is removed from a tissue, the remaining gases do not expand to fill the partial pressure vacated by the gas that was removed. The figure below shows total partial pressures for air breathing at 1 ATA from inspired gas to venous blood. Because of the decline in PO2 from alveoli to arterial blood, the total gas partial pressure in arterial blood during air breathing at 1 ATA is 752 mmHg, less than ambient pressure (760 mmHg). If PaO2 is lower than 95 mmHg (assumed in this example), then the total partial pressure in arterial blood will be less.

Would the alveolar-arterial PO2 difference (A-a)PO2 be normal in a healthy person breathing 100% oxygen; i.e., would alveolar and arterial PO2 both increase by the same amount?
No, the (A-a)PO2 will be greatly increased in healthy lungs breathing 100% O2.
The figure below shows inspired to venous blood partial pressures during O2 breathing at 1 ATA (100% O2 at sea level). In this example, it is assumed that all nitrogen, argon and other trace gasses have been washed out of the system. During O2 breathing, the ventilation/perfusion inequalities in lung have a much greater impact on PaO2 than during air breathing. Under optimal conditions during O2 breathing at 1 ATA, PaO2 would be about 500 mmHg while alveolar PO2 is about 663 mm Hg. (PAO2 = (760 - 47) - 40/.8 = 663). As blood moves through tissue, the same 4.5 mL O2/dL blood is extracted, and PO2 falls to 57 mmHg in venous blood.


I had a quick question on this image. Can you help me understand how the intrapleural pressure changes?I understand that the pressure inside the alveoli changes during inspiration because the volume increases, thus pressure has to decrease. During expiration the volume decreases thus the pressure increases.I don't know the changes in the intrapleaural pressure.

So the elastic recoil the same thing as transmural pressure. Should we just know that at rest our intrapleural pressure is at -5 and begin the cycle form there?Answer: The yellow arrow is meant to show the elastic recoil pressure of the lungs. I usually draw the arrow the other way since the lungs are "trying" to deflate (not get bigger). The alveolar pressure starts at zero (top figure). it is always the sum of intrapleural pressure (starts at - 5) and elastic recoil pressure (starts at +5). in drawing B, alveolar pressure is -1 because it equals -6.5 + 5.5 = -1. and so on for the other drawing.
Yes for the alveoli it is the same thing. For the airway transmural pressure is pressure inside minus pressure outside.
Question: Why does increasing the hematocrit (polycythemia, blood doping) cause the diffusion capacity to increase across the alveolar membrane?
Answer:
Khan video on diffusion
Not mentioned in the video is the influence of capillary volume and hemoglobin.
Capillary volume is part of the surface area component of the Fick Equation and is influence by factors such as exercise which increases diffusing capacity by increasing blood flow to the top of the lungs and thereby increasing capillary volume = surface area for diffusion.
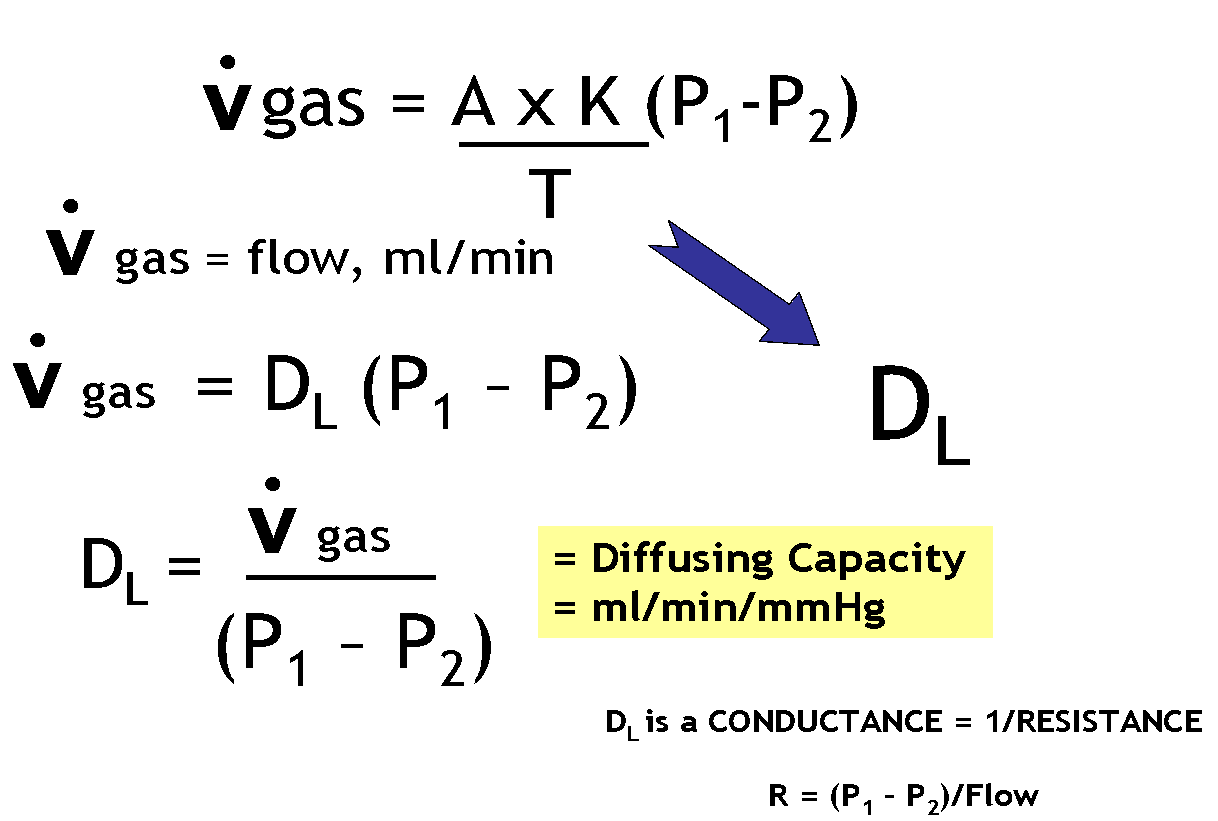
How is Diffusing Capacity Measured?

https://www.openanesthesia.org/pulm-diffusing-capacity/
Pulmonary diffusing capacity is often measured by Diffusion capacity of the Lungs for carbon monoxide (DLCO). In essence, this measures how much CO can pass from the alveoli to the blood in the pulmonary capillaries, thus giving clinicians the broader idea of how much inhaled gas can pass into the blood through the lungs.
While some state the “DLCO correlates with the total functioning surface area of the alveolar-capillary interface (Butterworth, et al),” Dr. McCormack notes, “Older textbooks suggest that thickening of the alveolar-capillary membrane (in interstitial lung disease) and loss of alveolar membrane surface area (in emphysema) are the primary causes of a low DLCO. However, subsequent experimental data suggest these and most other diseases that influence the DLCO do so by reducing the volume of red blood cells in the pulmonary capillaries” (McCormack). Regardless of theory, whether the surface area or the alveolar surface itself is modified or the volume of the blood in the pulmonary capillaries is modified, the DLCO reflects how much gas can be transferred to the blood via the lungs.
- Butterworth IV, JF, Mackey DC, Wasnick JD. Morgan & Mikhail’s Clinical Anesthesiology, 5th ed. New York, NY: McGraw Hill; 2013.
- McCormack, Meredith. “Diffusing Capacity for Carbon Monoxide.” Ed. James Stoller and Helen Hollingsworth. N.p., 14 Apr. 2015. Web.

Remember that most blood vessels only get sympathetic innervation - very few get cholinergic innervation.
Acetylcholine (ACh), whether released by cholinergic autonomic nerves or exogenously administered, binds to muscarinic receptors on the vascular endothelium (muscarinic receptors in coronary vessels), which stimulates the formation and release of NO as described above to produce vasodilation.


Production of nitric oxide (NO) by endothelial cells. NO is produced by the action of endothelial nitric oxide synthase (eNOS) on L-arginine. This reaction requires a number of cofactors, including tetrahydrobiopterin (BH4) and nicotinamide adenine dinucleotide phosphate (NADPH). Increased intercellular Ca++ in response to vasodilator agonists or shear stress displaces the inhibitor caveolin from calmodulin (CaM), activating eNOS. NO diffuses to vascular smooth muscle and causes relaxation by activating guanylate cyclase (GC), thereby increasing intracellular cyclic guanosine monophosphate (cGMP). Reprinted with permission from Behrendt D, Ganz P. Am J Cardiol. 2002;90(suppl):40L–48L.
I have a quick question about diastolic pressure. I was doing practice questions on Kaplan and one of the explanations said that mitral stenosis causes and increase in diastolic pressure and I'm a little confused on how. Would you be able to explain that to me?

Mitral stenosis (red PV loop in figure) impairs left ventricular filling so that there is a decrease in end-diastolic volume (preload). This leads to a decrease in stroke volume (reduced width of PV loop) by the Frank-Starling mechanism and a fall in cardiac output. Reduced ventricular filling and reduced aortic pressure decrease ventricular wall stress (afterload), which may result in a small decrease in ventricular end-systolic volume; however, this is not sufficient to offset the reduction in end-diastolic volume. Therefore, because end-diastolic volume decreases more than end-systolic volume decreases, the stroke volume (shown as the width of the loop) decreases.
I have a quick question about stroke work.
"A limitation of the relationship of stoke volume versus end-diastolic volume is that changes in either inotropic or afterload shifts the position of the curve.
if instead stroke work is plotted as a function of EVD, the position os the curve is independent of after load.
Stroke work is estimated as stroke volume times mean arterial blood pressure."
I understand the first part about shifting the curve for both inotropy and afterload, but i don’t understand what the graph for Stroke work is supposed to tell me. Its the same just with out after load? why do we remove after load?
I think the main factor is that stroke work incorporates pressure (afterload) so that there is no shift in the Starling curve due to changes in afterload (unlike the Starling curve that uses stroke volume vs end diastolic volume.

Why is the partial pressure of oxygen called oxygen tension? This might be trivial to understand but I feel that it might help me understanding ABGs at a deeper level. I’ve looked online at the Dalton and Henry equations from physics but it seems that the term “tension” is exclusively for medicine. Do you know why that is?
I have another question, if a person had a Right to left shunt, I know that the Qp/Qs ratio would be less than 1 because there will be greater systemic flow. I was thinking about the V/Q ratio and so would I be right to say that the V and Q would both be decreasing but the Q would have a greater affect and so the V/Q ration would be greater than 1 for a Right to left shunt?
I have a question regarding mitral stenosis. I'm trying to practice some pv loops and I came across the mitral stenosis one and I'm confused as to why End Diastolic Volume would decrease with mitral stenosis.
When I was trying to work through it, I figured that the Left atria would have an increase pressure because it has to work harder to get blood through the stenotic valve, hence there is a greater pressure gradient between LA and LV. I thought that that would increase filling time and increase end diastolic volume but that's not how it is and I'm confused why. Sorry for the long question but I appreciate all your help!!
Question: Also, just to clarify, force of contraction does not mean contractility or do they mean the same thing? If they don't, could you please explain it to me and how contractility is specific to Ca2+ levels?
Cardiac Inotropy (Contractility)
Effects of Inotropy on Frank-Starling Curves

Effects of Inotropy on Ventricular Pressure-Volume Loops

Factors Regulating Inotropy

Mechanisms of Inotropy
Question: Could you please explain to me how calcium channel blockers treat hypertension?
Answer. There are different classes of calcium channel blockers but in coronary and peripheral arterial smooth muscle and the heart, inhibition of Ca2+ channels blocks the entry of Ca2+ into cells and blunts the ability of Ca2+ to serve as an intracellular messenger. Therefore, calcium-channel blockers are smooth-muscle dilators and have a negative inotropic effect (decrease the force of contraction of the heart). both these effects cause a decrease in blood pressure.
Question: If there's an increase in insulin and K+ goes into the cell and there's hypokalemia which leads to a hyperpolarized resting membrane potential, why would the excitability increase? On your blog it says...
- Alkalosis leads to shift of K+ into cells = more negative Em (-80 to -90 mV) (by itself this might make the cell less excitable but the threshold for action potentials also become more negative.
- However, cells are more excitable because the rate of rise of phase 0 of the action potential (Vmax) is directly proportional to the value of the resting membrane potential at the onset of phase 0. This is because the membrane potential at the onset of depolarization determines the number of sodium channels activated during depolarization, which in turn determines the magnitude of the inward sodium current and the Vmax of the action potential.

- Shift of H+ out of cells leads to increased protein negatively charged binding (Pr-) sites inside cells and in plasma = increased Ca++ binding to these negative sites = decreased contractility
- Net effect = increased excitability, cardiac arrhythmias; muscle weakness

Asthma
Question
When as pt has and asthma attack due to it being an obstructive problem , I thought the pt would be hypercapnic .. The PBL had the ABGS that showed hypocapnic. I dont follow the rational. Also, when A beta 2 agonist is given , does it initially cause hypoxemia because the vessels were vasodilated and when the beta agonist was given it caused vasoconstriction and this caused a Ventilation/respiration mismatch?
Potassium
I'm finding a few little discrepancies between the currents discussed in class and those in Physiology by Costanzo, and was wondering what to go by. For example, regarding Phase 3, Costanzo never mentions an I(k1), only an I(k), but we had talked about both in class for this phase. Also for phase 4, Costanzo says for action potentials in the ventricles and atria, there is an outward I(k1) current and inward i(Na) and i(Ca-T), but in class we said it was an outward i(k) and inward i(Ca-T) and i(f). The only time Costanzo mentions i(f) is with the SA node action potential. Are these differences really important?
Regarding the question which begins "This EKG was obtained from a 77 year old woman who was recently admitted to the Coronary Care Unit for an acute inferior myocardial infarction...." the question is accompanied by an EKG which shows a narrow complex tachycardia without discernable p-waves. It appears to have left axis deviation (Up in I, Down in aVF). The V leads are not concordant. The answer is keyed as V-tach, not SVT. If this is VT, what did I miss?
Answer 1
I can see why this is confusing. Looking at the QRS complexes they are not as wide and bizarre looking as we usually see in VT although they are definitely widened a little. The thing that really makes it VT is the AV dissociation with the appearance of p waves independent of the QRS complexes (indicated by the arrows). You cannot have AV dissociation with SVT because the atria (and SA node) are depolarized at the same rate as the ventricle, around 150 times/minute. That means that the SA node would never get a chance to depolarize normally and cause a p-wave.
Answer 2
In short, SVT is an "above the ventricle" arrhythmia. Typical SVTs include: A-Fib, A-Flutter, AVRT, or AVNRT. VT is a ventricular initiated arrhythmia. This rhythm is often a medical emergency as it may digress to VF and significantly compromise systemic perfusion.
On the ECG one could often distinguish between the two by looking at the width of the QRS complexes. VT will be very broad suggesting ventricular delay, SVT tends to be narrow.
In the example question given the QRS complexes appear to be >100 msec (wide) but this is not clear given no little box detail and no print out of intervals.
Rho Kinase
I just have a quick question I'm having a hard time answering. Does the RhoA/ROCK pathway stimulate or inhibit the MLCP pathway?
Answer
Activation of the RhoA-ROK pathway mediates increased smooth muscle Ca2+ sensitivity by inhibition of MLCP, and provides a major contribution to receptor-mediated Ca2+-sensitization of smooth muscle. Such inhibition of MLCP results in accumulation of phosphorylated MLC and thus greater contraction for a given [Ca2+]i.
Myocyte Action Potentials and Ion Conductance
summative 2 had MCQ on this. Figure was flawed. Here is an accurate figure

Answer
"I think that the scale on the "Ion Conductivity" axis is incorrect. At the end of phase 4, all conductances shouldn't be the same. (It looks sort of like the figure is showing current rather than conductance.) In a graph of conductance, gK should be high, it falls during phase 0 and gradually rises again during phase 2 and 3. gNa is transient during phase 0 as shown and L-type gCa is at a peak during phase 2. Further confusing is the time scale, which is compressed with respect to that of the action potential. The figure below from makes this point. (This image is fromhttp://www.cvpharmacology.com/antiarrhy/cardiac_action_potentials.htm, which along with it's links, provides a pretty clear description of the cardiac action potential.)
4 comments:
Thank you for the good writeup. It in truth used to be a enjoyment account it.
Glance advanced to far brought agreeable from you!
However, how can we keep in touch?
Hi there, I enjoy reading all of your article.
I like to write a little comment to support you.
thanks
test
Post a Comment